

Аномалия Киари представляет собой структурный дефект мозжечка.. Это часть мозга, отвечающая за равновесие..
в обычной ситуации, мозжечок и часть ствола мозга покоятся в пространстве, расположенном над отверстием, которое называется большим затылочным отверстием.. Когда возникает порок, мозжечок расположен ниже большого затылочного отверстия, увеличивая давление на нервы и ткани в этой области.

Ученые считали, что аномалия Киари возникает в 1 десятилетие 1,000 рождения, но новые методы диагностической визуализации, как КТ (TC) и магнитно-резонансная томография (IRM), предположить, что это состояние гораздо более распространено.
Точные оценки сделать сложно, потому что некоторые дети рождаются с этим заболеванием, и у них никогда не проявляются симптомы или симптомы развиваются в подростковом или взрослом возрасте. В равной степени, Доказано, что аномалия Киари поражает женщин чаще, чем мужчин..
Существует четыре типа синдрома Арнольда-Киари.. Это редкие заболевания, но первый (тип I) Это наиболее распространенный и менее серьезный, чем остальные.
Другие заболевания, связанные с этим пороком развития:: гидроцефалия, расщелина позвоночника, сирингомиелия и синдром фиксированного пуповины. Его часто можно увидеть у подростков или взрослых..
Показатель
Как классифицируют мальформацию Киари??
В конце 19 века, патологоанатом, а также профессор Ганс Киари, обнаружены аномалии головного мозга, между местом соединения черепа и позвоночника. Они были ранжированы в порядке серьезности., как тип I, II, III и IV, тем не менее, пороки развития I типа являются наиболее распространенными.
Мальформация Киари. Тип I.
Это то, что происходит во время развития плода.. Характеризуется смещением миндалин мозжечка вниз в шейный отдел позвоночного канала., около, четыре миллиметра. Из-за этого смещения блокируется нормальная циркуляция спинномозговой жидкости. (LCR) между позвоночным каналом и внутричерепным пространством.
Аномалии, присутствующие при аномалии Киари
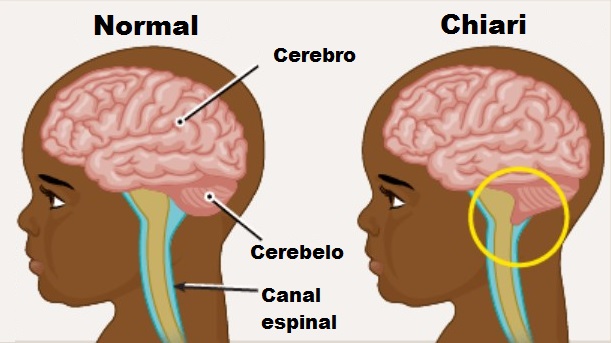

Тип I.
При аномалии Киари I типа, аномалии наблюдаются в основании черепа и позвоночнике между 30 а также 50 процент пациентов. Это включает:
- Сдавление основания черепа и, как следствие, сдавление ствола головного мозга..
- Костное прикрепление позвонка С1 к основанию черепа
- Частичное сращение С1 и С2 позвонков позвоночника
- Врожденное сращение или слияние в области шеи с возможной ассоциированной мальформацией верхних уровней шейного отдела позвоночника. (Деформидад де Клиппель-Фейль)
- Скрытая расщелина позвоночника
- Присутствие сколиоз, особенно у детей с незрелым позвоночником
Тип II
При мальформации Киари II типа, наблюдается смещение ниже мозгового вещества, мозжечок и четвертый желудочек в шейном отделе позвоночного канала. Обычно возникает у пациентов с миеломенингоцеле (врожденное состояние во время развития плода, где спинной мозг и позвоночник не закрываются должным образом).
Тип III
При аномалии Киари III типа, дизрафия наблюдается в части мозжечка и / или ствола мозга. тем не менее, эта аномалия встречается очень редко. Отмечается высокая ранняя смертность младенцев, страдающих им, а те, кому удается выжить, имеют выраженный неврологический дефицит..
В качестве лечения предпринимается раннее хирургическое закрытие дефекта.. Порок развития может сопровождаться тяжелыми врожденными дефектами., что может потребовать обширного лечения. Но, как мы упоминали выше, у детей могут быть опасные для жизни осложнения.
Тип IV
Мальформация Киари IV типа — самая тяжелая форма и самая редкая.. Это происходит, когда мозжечок не развивается нормально.. Дети с этим пороком развития, вряд ли перерастет детство.
Симптомы мальформации Киари

Тип I.
В некоторых случаях симптомы отсутствуют. Многие симптомы связаны с образованием сиринкса.. тем не менее, когда они проявляются, они могут возникать отдельно или в сочетании со следующими:
- Головная боль, возникающая в основании черепа, ухудшается при чихании или кашле
- Головокружение и проблемы с равновесием
- Мышечная слабость в руках и кистях. В других случаях наблюдается мышечное напряжение.
- Апноэ во сне
- Визуальные проблемы, Какие: повышенная чувствительность к свету, двойное или нечеткое зрение
Тип II
Симптомы связаны с миеломенингоцеле или гидроцефалией., Такие как:
- Периоды апноэ или изменение нормального ритма дыхания
- Тошнота
- Слабость и потеря силы в руках
- непроизвольные движения глаз
Тип III
Симптомы появляются в детском возрасте и сопровождаются опасными для жизни осложнениями.. Присутствуют те же симптомы порока развития II типа., но с неврологическими дефектами, такими как умственная отсталость и судороги.
Тип IV
- Сколиоз
- потеря чувствительности, включая горячее и холодное
- сильная общая боль, особенно в голове
- Потеря контроля над кишечником и мочевым пузырем
- Сильная мышечная слабость и спастичность
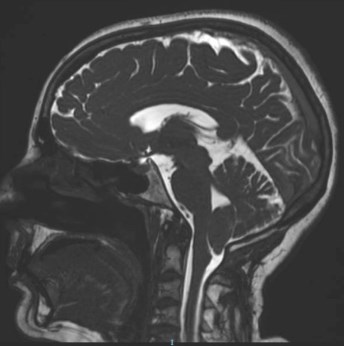

Для диагностики аномалии Киари существует несколько тестов, которые могут определить:, даже какой это тип. давайте посмотрим, какие они:
Магнитно-резонансная томография (IRM). Это первый диагностический тест, поскольку он дает трехмерные изображения.. Обеспечивает точное изображение мозжечка, спинной и головной мозг, с помощью этого теста можно определить объем пороков развития и даже различить прогрессирование.
Компьютерная томография (ТС или ТАС). Этот тест использует рентгеновские лучи для создания изображения. С его помощью можно определить размер желудочков головного мозга и показать закупорку, если бы были. Костные аномалии в основании головного мозга и цервикальном канале могут быть оценены.
Миограмма. Процесс заключается во введении контрастного вещества, сделать рентген позвоночного канала. Цель состоит в том, чтобы показать давление, оказываемое пороком развития на спинной мозг или нервы.. это редкий тест.
анализ сна. Больного усыпляют в специальной комнате., где можно контролировать дыхательную активность, уровни оксигенации и возможные судороги для демонстрации апноэ во сне.
тест на глотание. С помощью рентгена наблюдают за прохождением пищи через глотку, чтобы определить любую аномалию, которая предполагает дисфункцию нижних отделов ствола мозга..

Мальформация Киари имеет множество различных причин.. В большинстве случаев устанавливается врожденная причина.. Во время внутриутробного развития в головном и спинном мозге выявляются структурные дефекты.. Это может быть вызвано генетическими мутациями или неправильным питанием с дефицитом витаминов и питательных веществ во время беременности..
Лечение аномалии Киари
Лечение мальформации Киари зависит от точного типа мальформации., а также прогрессирование и симптомы, представленные.
При бессимптомной мальформации лечение не проводится. (обычно тип I). Если порок развития симптоматический, лечение можно продолжать до тех пор, пока установлено, что гидроцефалия не вызывает осложнений.
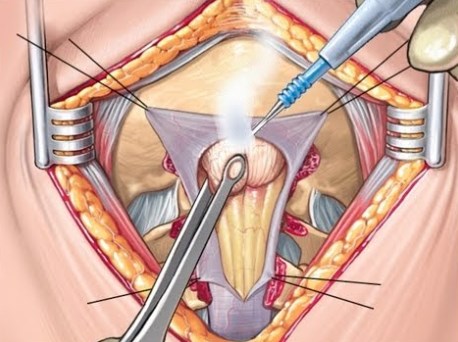

Хирургическое лечение зависит от степени компрессии или других аномалий.. Мальформацию Киари I можно лечить хирургическим путем с локальной декомпрессией перекрывающихся костей и высвобождением твердой мозговой оболочки.. Некоторым пациентам потребуется спондилодез шейного отдела позвоночника..
Аналогичным образом лечится декомпрессия мальформации Киари II., но ограничивается декомпрессией тканей позвоночного канала. Эта декомпрессия проводится под общим наркозом..
Эта операция направлена на эффективную декомпрессию нервной ткани и восстановление нормального потока спинномозговой жидкости вокруг и позади мозжечка..
Хирургия не является гарантией успеха для каждого человека.. Повреждение нерва, вызванное пороком развития, не может быть устранено.. Некоторым пациентам требуется более одной операции, а в некоторых случаях облегчение симптомов не достигается..